Síndrome de Lynch (HNPCC - câncer colorretal hereditário não polipose) - manifestações clínicas e diagnóstico
●O câncer colorretal hereditário não polipose refere-se a pacientes e/ou famílias que preenchem os critérios de Amsterdam (tabela 1). Uma parte desses pacientes terá síndrome de Lynch em testes moleculares germinativos. (Consulte 'Critérios baseados no histórico da família' abaixo.)
●A síndrome de Lynch refere-se a pacientes e famílias com uma mutação germinativa em um dos genes de reparo de incompatibilidade de DNA (MLH1, MSH2, MSH6, PMS2) ou no gene EPCAM.
Epidemiologia da síndrome de Lynch
Genética da síndrome de Lynch
Manifestações colônicas da síndrome de Lynch
Manifestações extra-colônicas da síndrome de Lynch
Identificação de indivíduos em risco para a síndrome de Lynch
Características do tumor na síndrome de Lynch
Critérios de Amsterdã na síndrome de Lynch
Critérios de Bethesda revisados na síndrome de Lynch
Abordagem diagnóstica na suspeita da síndrome de Lynch
Teste de linha germinativa da síndrome de Lynch
Interpretação do teste germinativo na síndrome de Lynch
Indicações para teste de tumor na síndrome de Lynch
Teste de instabilidade de microssatélites na síndrome de Lynch
Teste imuno-histoquímico (IHC) na síndrome de Lynch
Diagnóstico diferencial da síndrome de Lynch
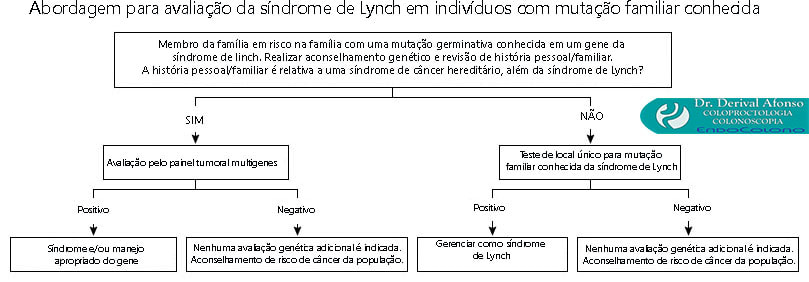
Avaliação genética de familiares para a síndrome de Lynch
RESUMO E RECOMENDAÇÕES PARA A SÍNDROME DE LYNCH
Os genes MMR que estão associados à síndrome de Lynch incluem:
●MLH1 (MutL homólogo 1), que está localizado no cromossomo 3p22.2
●MSH2 (MutS homólogo 2), que está localizado no cromossomo 2p21-16
●MSH6 (MutS homólogo 6), que está localizado no cromossomo 2p16.3
●PMS2 (segregação pós-meiótica 2), que está localizado no cromossomo 7p22.1
Os riscos de CCR e câncer endometrial são semelhantes em indivíduos com mutações MLH1 e MSH2, mas os riscos para câncer urotelial e manifestações cutâneas da síndrome de Lynch são maiores em portadores da mutação MSH2.
Embora a idade de início varie de acordo com o genótipo, o CCR na síndrome de Lynch ocorre frequentemente em uma idade mais jovem em comparação com o CCR esporádico. No entanto, deve-se notar também que muitos CCRs em pacientes com síndrome de Lynch ocorrerão acima de 50 anos.
Indivíduos com síndrome de Lynch apresentam risco aumentado para CCRs síncronos (simultâneos) e metacrônicos (épocas diferentes). Aproximadamente 7 por cento das pessoas com síndrome de Lynch têm mais de um câncer no momento do diagnóstico. Em indivíduos com síndrome de Lynch que foram submetidos apenas a uma ressecção segmentar para o primeiro câncer de cólon, 16% desenvolveram um CCR metacrônico em 10 anos, 41% em 20 anos e 62% em 30 anos. Da mesma forma, em indivíduos com síndrome de Lynch que foram submetidos à cirurgia para o primeiro câncer retal, 19% desenvolveram um CCR metacrônico em 10 anos, 47% em 20 anos e 69% em 30 anos.
Os CCRs na síndrome de Lynch diferem dos CCRs esporádicos, pois são predominantemente localizados no lado direito. Embora se acredite que a maioria dos CCRs associados a Lynch evoluam de adenomas, os adenomas tendem a ser maiores, mais planos, mais frequentemente proximais e são mais propensos a ter displasia de alto grau e/ou histologia de vilosidades em comparação com adenomas esporádicos.
A sequência adenoma-carcinoma também pode progredir mais rapidamente na síndrome de Lynch em comparação com o CCR esporádico (35 meses versus 10 a 15 anos), mas o câncer pode se desenvolver diretamente de criptas microscópicas da mucosa do cólon. No entanto, a sobrevida global de 10 anos do CCR na síndrome de Lynch é alta (88% para câncer de cólon e 70% para câncer reto-sigmóide).
Crescente evidências sugere que homens com síndrome de Lynch estão em maior risco de câncer de próstata, que pode chegar a 30% de risco ao longo da vida. Um risco aumentado de câncer de mama feminino também foi relatado em vários estudos entre mulheres com síndrome de Lynch, no entanto, outros não detectaram um risco aumentado. Vários outros cânceres foram relatados em indivíduos com síndrome de Lynch, mas não está claro se a incidência desses cânceres está acima da população geral.
A triagem universal de câncer de endométrio e CCR para instabilidade de microssatélites (MSI) melhorou a identificação da síndrome de Lynch entre pacientes com câncer. Dados de estudos também sugerem que mutações da síndrome de Lynch, principalmente MSH6 e PMS2, também estão presentes em uma pequena porcentagem de pacientes com tumores estáveis de microssatélites (MSS).
Critérios baseados no histórico familiar na síndrome de Lynch
Vários critérios baseados no histórico familiar têm sido usados para identificar indivíduos em risco de síndrome de Lynch. Eles têm sensibilidade limitada para identificação de pacientes com síndrome de Lynch, mas são úteis para orientar a abordagem da avaliação genética.
- Deve haver pelo menos três parentes com qualquer câncer associado à síndrome de Lynch (câncer colorretal, câncer de endométrio, intestino delgado, ureter ou pelve renal)
- Um deve ser parente de primeiro grau dos outros dois
- Pelo menos duas gerações sucessivas devem ser afetadas
- Pelo menos um deve ser diagnosticado antes dos 50 anos
- A polipose adenomatosa familiar deve ser excluída no(s) caso(s) de câncer colorretal, se houver
- Tumores devem ser verificados por exame patológico
1. Câncer colorretal diagnosticado em paciente com menos de 50 anos de idade.
2. Presença de tumores colorretais sincrônicos, metacrônicos ou outros associados ao HNPCC *, independentemente da idade.
3. Câncer colorretal com histologia semelhante a MSI-H ¶ Δ diagnosticado em paciente com menos de 60 anos de idade ◊.
4. Câncer colorretal diagnosticado em paciente com um ou mais parentes de primeiro grau com tumor relacionado ao HNPCC, sendo um dos cânceres diagnosticado com menos de 50 anos.
5. Câncer colorretal diagnosticado em paciente com dois ou mais parentes de primeiro ou segundo grau com tumores relacionados ao HNPCC, independentemente da idade.
HNPCC: câncer colorretal hereditário nãom polipose; MSI-H: instabilidade de microssatélites alta.
* Tumores relacionados ao HNPCC incluem tumores colorretais, endometriais, gástricos, ovarianos, pâncreas, ureter e pelve renal, trato biliar e cérebro (geralmente glioblastoma como visto na síndrome de Turcot), adenomas de glândulas sebáceas e ceratocantomas e carcinoma do intestino delgado.
¶ MSI-H em tumores refere-se a alterações em dois ou mais dos cinco painéis de marcadores microssatélites recomendados pelo National Cancer Institute.
Δ Presença de linfócitos infiltrantes do tumor. Reação linfocítica semelhante a Crohn, diferenciação mucinosa/em anel de sinete ou padrão de crescimento medular.
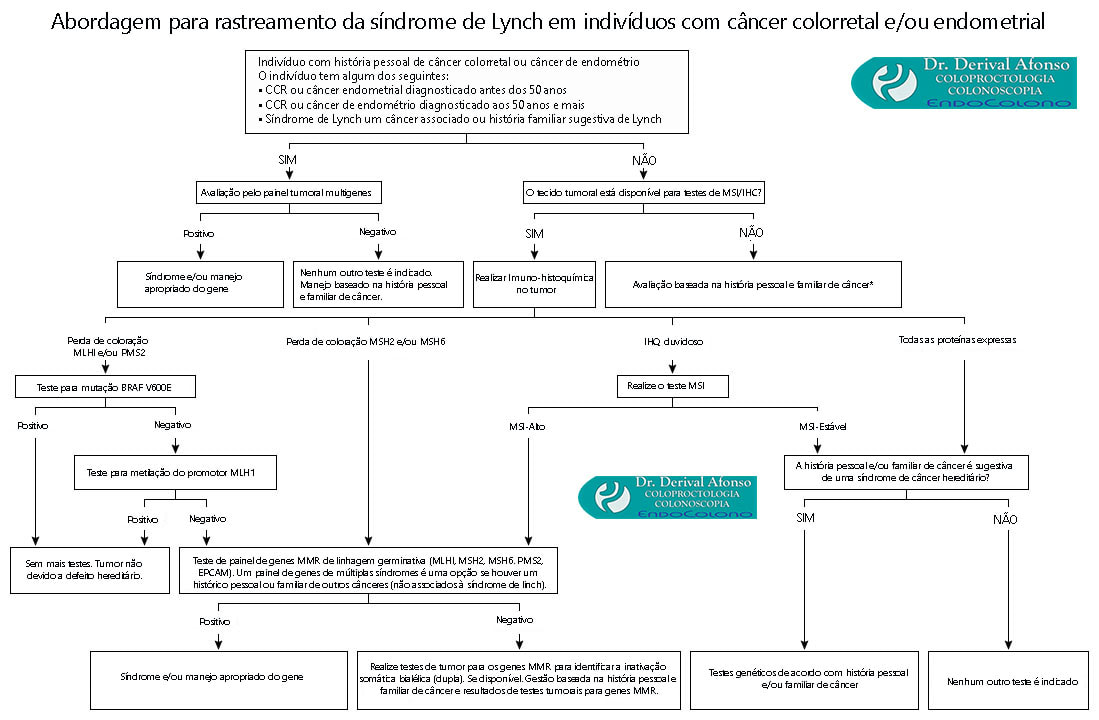
A abordagem para avaliação da síndrome de Lynch varia com base na história pessoal ou familiar de câncer associado a Lynch, incluindo os tipos de câncer e a idade de início. Idealmente, a avaliação genética para a síndrome de Lynch deve começar com o paciente afetado com um câncer da síndrome de Lynch.
A avaliação genética para a síndrome de Lynch está indicada nas seguintes situações:
●CCR ou câncer de endométrio antes dos 50 anos;
●CCR ou câncer de endométrio diagnosticado com idade > 50 anos com história pessoal e familiar adicional sugestiva de síndrome de Lynch;
●Identificação de uma variante patogênica de MMR no teste de tumor somático em qualquer tipo de tumor;
●Indivíduos não afetados (sem câncer) com um dos seguintes:
•História familiar de câncer que atende aos critérios Amsterdam I ou II ou diretrizes revisadas da Bethesda;
•Parente de primeiro ou segundo grau daqueles com mutação genética MMR/EPCAM conhecida.
A triagem genética baseada nas características do tumor (mucinosos, células em anel de sinete ou tipo histológico medular) para a síndrome de Lynch com teste MSI ou imuno-histoquímica deve ser realizada:
●CCR aos 50 anos ou mais;
●Câncer de endométrio em qualquer idade.
Indivíduos com tumores MSI-high (MSI-H) ou perda de expressão de uma proteína MMR em IHC requerem testes adicionais com base no padrão de perda de expressão de uma proteína MMR. Se o teste no tumor não for viável e a suspeita clínica de risco de câncer hereditário devido ao histórico familiar for forte ou indisponível (por exemplo, devido à adoção, doação de esperma etc.), os pacientes devem receber testes germinativos.
●Pacientes diagnosticados com CCR ou câncer de endométrio antes dos 50 anos de idade;
●Indivíduos com CCR ou câncer de endométrio diagnosticados com idade > 50 anos com história pessoal e familiar adicional sugestiva de síndrome de Lynch;
●Identificação de uma variante patogênica de MMR no teste de tumor somático em qualquer tipo de tumor;
●Se o teste do tumor não for viável (indivíduo não afetado) e a suspeita clínica de risco de câncer hereditário devido ao histórico familiar for forte ou totalmente indisponível (por exemplo, devido à adoção, doação de esperma etc.).
Sugere-se a avaliação genética dos pacientes com suspeita de síndrome CCR hereditária com testes de painel multigênico para avaliar todos os genes de alto risco potenciais ou suspeitos simultaneamente. Essa abordagem avalia aproximadamente 10 a 15 genes de alto risco para o CCR (incluindo os genes MMR da síndrome de Lynch) simultaneamente, embora isso varie entre os laboratórios clínicos. Os custos associados à avaliação de risco genético e testes genéticos germinativos caíram drasticamente, aumentando o acesso aos testes para a síndrome de Lynch e outras síndromes hereditárias e genes de alto risco.
O teste de painel multigênico é o meio mais eficiente em termos de tempo e custo para avaliar um paciente com CCR de início precoce (<50 anos) para genes de penetrância alta e moderada que podem ser identificados. Estudos demonstraram que, embora um número substancial de pacientes com CCR de início precoce (antes dos 50 anos) tenha síndrome de Lynch (aproximadamente 8%), muitos outros carregam mutações em outros CCR associados e não associados ao CCR devido a outros genes clinicamente acionáveis.
Em pacientes com 50 anos ou mais com um CCR deficiente de MMR por triagem MSI ou IHC, uma abordagem direcionada para testar a síndrome de Lynch também pode ser realizada se a triagem tumoral universal detectou deficiência de MMR e levantou a suspeita de síndrome de Lynch. No entanto, se houver histórico pessoal ou familiar de outros cânceres (não associados à síndrome de Lynch), o teste de painel multigênico deve ser realizado.
●Se uma variante da doença foi identificada em uma família e o indivíduo testado não possui essa variante, isso exclui o diagnóstico de síndrome de Lynch.
●Uma variante de significância desconhecida (VSD) em um indivíduo de risco é um achado inconclusivo e não estabelece a presença ou ausência de risco no indivíduo testado. No entanto, o sequenciamento de DNA normal (sem VSD) oferece algum grau de garantia de que os genes de risco hereditário testados parecem ter sequências normais (não mutadas) quando comparados com as sequências de referência usadas pelo laboratório que realiza o teste. Em indivíduos com VSD, a interpretação atualizada da patogenicidade deve ser obtida periodicamente.
O teste de tumores para evidência de MMR de DNA deficiente tem sido usado para identificar indivíduos em risco de síndrome de Lynch.
Embora, alguns indicam a triagem genética no tumor com testes MSI ou IHC para a síndrome de Lynch em todos os CCRs, independentemente da idade, porque a sensibilidade é ligeiramente maior para a identificação da síndrome de Lynch em comparação com outras estratégias e demonstrou ser custo-efetiva.
A sensibilidade e especificidade do teste MSI para a síndrome de Lynch são de aproximadamente 85 e 90 por cento, respectivamente. MSI não é específico para a síndrome de Lynch, e aproximadamente 15 por cento de todos os CCRs esporádicos demonstram MSI devido à hipermetilação de MLH1, portanto, quando presente deve-se fazer teste germinativo.
O teste IHQ no tecido tumoral para falta de expressão de proteínas MMR tem sensibilidade e especificidade de 83 e 89 por cento, respectivamente. A IHQ geralmente está mais disponível porque pode ser realizada em pequenas biópsias, é barata e tem o valor na identificação de qual dos genes MMR pode estar causando o tumor.
A sensibilidade da IHQ para a síndrome de Lynch pode estar diminuída no tecido tumoral retal que foi previamente irradiado. Portanto, a realização de IHQ em uma biópsia pré-tratamento é ideal.
A IHQ em outros tumores da síndrome de Lynch foi estudada com menos rigor e, embora estudos tumorais anormais possam ser indicativos de síndrome de Lynch, estudos tumorais normais não excluem necessariamente a síndrome de Lynch. Os resultados da IHQ nestes tumores devem ser avaliados à luz da história familiar e pessoal de tumores.
●MSI-H ou perda de expressão de uma proteína MMR na IHQ – A decisão entre a realização do painel multigênico ou testes mais seletivos para a síndrome de Lynch é baseada no padrão de perda de IHQ.
•Perda de MLH1/PMS2 – CCRs que mostram perda de MLH1/PMS2 na IHQ podem ser testados para a mutação BRAF V600E comum para ajudar a determinar se a perda de expressão de MLH1/PMS2 por IHQ é devido à síndrome de Lynch ou hipermetilação do promotor MLH1.
•Perda de expressão de MSH2/MSH6 – Em indivíduos com perda de expressão de MSH2/MSH6, a decisão de buscar testes de painel multigênico versus testes focados apenas no diagnóstico da síndrome de Lynch deve ser baseada em um histórico familiar abrangente.
À medida que o teste genético em tumores se tornou clinicamente disponível, deve ser oferecido àqueles indivíduos que se enquadram nesse perfil, pois a comprovação da causa somática evitará que sejam erroneamente rotulados como síndrome de Lynch, com toda a triagem e riscos inerentes.
●Síndrome da deficiência de reparo de incompatibilidade constitucional (CMMR-D) – Refere-se a pacientes e/ou famílias com mutações bialélicas germinativas de um gene MMR de DNA. Em contraste com a síndrome de Lynch, na qual os cânceres ocorrem na quinta ou sexta década de vida, portadores de mutações homozigóticas ou heterozigotas compostas de mutações MLH1, MSH2, MSH6 ou PMS2 geralmente desenvolvem malignidades associadas a Lynch, malignidades hematológicas e cerebrais e sarcomas durante infância, muitas vezes nas primeiras décadas de vida.
●Câncer Colorretal Familiar Tipo X (FCCTX) – Refere-se a pacientes e/ou famílias que atendem aos critérios de Amsterdam I, mas quando os tumores são testados, não apresentam MSI que é característico da síndrome de Lynch. Pacientes com FCCTX também não parecem ter um risco aumentado de câncer endometrial ou outros cânceres associados a Lynch. Esses indivíduos devem ser submetidos a testes de painel multigênico para descartar causas genéticas raras de CCR familiar.
●Síndrome de Lynch Clínica – A síndrome de Lynch clínica descreve indivíduos raros sem explicação clara para seu tumor deficiente em MMR. Os indivíduos têm um tumor MSI-H, mas não terão uma mutação germinativa da síndrome de Lynch, não terão hipermetilação do promotor MLH1 e não terão evidência de inativação somática bialélica de um dos genes da síndrome de Lynch. Esses indivíduos e famílias são tratados clinicamente como síndrome de Lynch e/ou têm manejo fortemente guiado pela história pessoal/familiar.
●Polipose adenomatosa familiar atenuada (PAFA) e polipose associada a MUTYH (MAP) – Indivíduos com PAFA e MAP e alguns adenomas colorretais podem ser difíceis de distinguir clinicamente da síndrome de Lynch. Apenas testes genéticos podem distinguir definitivamente entre Mapa, MAP e síndrome de Lynch, embora um padrão autossômico dominante de herança CCR torne a MAP improvável. A PAFA é caracterizada por mutações germinativas no gene APC e indivíduos com MAP apresentam mutações bialélicas nos genes MUTYH .
●O risco ao longo da vida de CCR até a idade de 70 anos na síndrome de Lynch é de 10 a 90%, dependendo do sexo do paciente, do gene MMR mutado e da penetrância do gene na família. Indivíduos com síndrome de Lynch também apresentam risco aumentado de câncer de endométrio e várias outras malignidades, incluindo câncer de ovário, estômago e sistema geniturinário e, mais raramente, intestino delgado, ducto biliar, pâncreas, pele (neoplasias sebáceas) e cérebro. tumores (gliomas). Os portadores de PMS2, em particular, têm um risco de câncer ao longo da vida menor do que os outros genes da síndrome de Lynch.
●Os CCRs na síndrome de Lynch diferem dos CCRs esporádicos, pois são predominantemente localizados no lado direito. Embora os CCRs pareçam evoluir de adenomas, os adenomas tendem a ser maiores, menos elevados (planos), mais frequentemente proximais (cólon direito) e mais comumente apresentam displasia de alto grau e/ou histologia vilosa em comparação com adenomas esporádicos. A sequência adenoma-carcinoma também progride muito mais rapidamente na síndrome de Lynch em comparação com o CCR esporádico. Indivíduos com síndrome de Lynch estão em risco aumentado para CCRs síncronos e metacrônicos.
●Os tumores na síndrome de Lynch geralmente mostram instabilidade de microssatélites (MSI) e perda de coloração das proteínas MMR no teste de imuno-histoquímico (IHQ). Em comparação com os CCRs esporádicos, eles são mais frequentemente mucinosos, tipo histológico de células em anel de sinete ou medular, pouco diferenciados e têm um infiltrado linfocítico vivo ou são margeados por uma reação linfoide produtora de centro germinativo semelhante a Crohn.
●Vários critérios clínico-patológicos (por exemplo, critérios de Amsterdã e diretrizes revisadas de Bethesda) foram historicamente usados para identificar indivíduos em risco de síndrome de Lynch ou elegíveis para teste MSI, mas são menos relevantes para a prática clínica devido à baixa sensibilidade para a síndrome de Lynch. A triagem universal de câncer de endométrio e CCR por testes MSI ou IHC de casos incidentes está sendo cada vez mais realizada rotineiramente para a identificação da síndrome de Lynch. (Consulte 'Identificação de indivíduos em risco de síndrome de Lynch' acima.)
●A síndrome de Lynch deve ser suspeitada em pacientes com CCR sincrônico ou metacrônico, CCR ou câncer de endométrio antes dos 50 anos de idade, múltiplos cânceres associados a Lynch (p. e em casos de agrupamento familiar de cânceres associados a Lynch. A síndrome de Lynch também deve ser suspeitada em qualquer tumor que tenha MMR deficiente nos testes de MSI ou IHC. (Consulte 'Abordagem de diagnóstico' acima.)
●A abordagem para avaliação da síndrome de Lynch varia de acordo com o histórico pessoal ou familiar de câncer associado a Lynch, o tipo de câncer e a idade de início. Idealmente, a avaliação genética para a síndrome de Lynch deve começar com um paciente afetado com um câncer da síndrome de Lynch.
●Começamos com a avaliação genética da linhagem germinativa para a síndrome de Lynch em indivíduos com um dos seguintes (algoritmo 1ealgoritmo 2) (consulte 'Teste de linha germinativa' acima):
•CCR ou câncer de endométrio antes dos 50 anos
•CCR ou câncer de endométrio diagnosticado com idade > 50 anos com história pessoal e familiar adicional sugestiva de síndrome de Lynch
•Indivíduos não afetados (sem câncer) com um dos seguintes:
-≥2,5 por cento de chance de uma mutação do gene MMR por modelos de previsão (consulte 'Modelos de previsão clínica' acima)
-História familiar de câncer que atende aos critérios Amsterdam I ou II ou diretrizes revisadas da Bethesda (consulte 'Critérios baseados na história familiar' acima)
-Parente de primeiro ou segundo grau daqueles com mutação genética MMR/ EPCAM conhecida
Triagem genética baseada em tumor para síndrome de Lynch com teste MSI ou IHC em indivíduos com um dos seguintes (consulte 'Características do tumor' acima):
•CCR aos 50 anos ou mais
•Câncer de endométrio aos 50 anos ou mais
Indivíduos com tumores de alto MSI ou perda de expressão de uma proteína MMR em IHC requerem testes adicionais com base no padrão de perda de expressão de uma proteína MMR.
As informações contidas neste artigo são apenas para fins educacionais e não devem ser usadas para diagnóstico ou para orientar o tratamento sem o parecer de um profissional de saúde. Qualquer leitor que está preocupado com sua saúde deve entrar em contato com um médico para aconselhamento.