
PÓLIPO COLORRETAL
Introdução e Sintomas.
Diagnóstico dos pólipos colorretais.
Classificação Macroscópica e Microscópica (histológica).
Sequência Adenoma-Carcinoma e Serrilhado-Carcinoma.
Carcinoma “de novo”.
Síndromes hereditárias associadas ao câncer colorretal.
Polipose Adenomatosa Familiar ou FAP
Câncer Colorretal Hereditário Sem Polipose (HNPCC).
Outras Poliposes Colônicas.
Tratamento dos pólipos colorretais.
----Estudo dos pólipos colorretais pela colonoscopia.
----Técnica de retirada dos pólipos colorretais pela colonoscopia.
----Complicações da polipectomia e mucosectomia do cólon.
Pólipo com Câncer Colorretal (maligno).
Seguimento após a retirada de pólipo colorretal (seguimento pós-polipectomia).
Quando parar com a colonsocopia de prevenção?
Recomendações em casos especiais
Preparo para a colonoscopia.
➀ Sangrar, saindo pelo ânus sangue vivo (hematoquezia) ou escuro (melena) podendo causar anemia por deficiência de ferro;
➁ Produzir muco (catarro), saindo pelo ânus isoladamente ou misturado ao sangue e quando abundante pode causar desidratação com diminuição do potássio;
➂ Quando grandes podem obstruir parcialmente (causa dor e inchaço abdominal) ou totalmente (abdome agudo obstrutivo) o intestino e/ou causar alternância do hábito intestinal (ora diarreia ora constipação).
➀ Os pólipos são comuns, e estão presentes em cerca de 30% dos adultos com 45 anos ou mais;
➁ Nem todos os tipos de pólipo têm a capacidade de evoluir para o câncer de intestino;
➂ São necessários alguns anos (10 a 15 anos dependendo do tamanho e da classificação) para que o pólipo se transforme em câncer, já que o mesmo é um tumor benigno;
➃ A maioria dos pólipos pode ser retirada com segurança pela colonoscopia.
➁ Através da colonoscopia de prevenção em pacientes assintomáticos.
B- Quem já teve câncer colorretal
C- Mulheres com câncer de mama, ovário ou útero
D- Portadores da Doença de Crohn e Retocolite Ulcerativa

|
Lesões Polipoides
ᴥ» Tipo Ip - lesão pediculada ᴥ» Tipo Is - lesão séssil Lesões Superficiais do Cólon
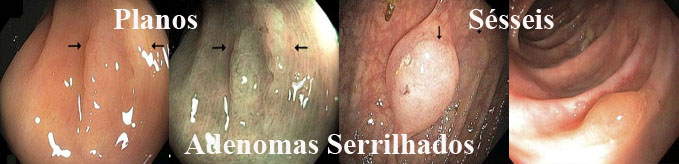
ᴥ» Tipo IIa - lesão levemente elevada; ᴥ» Tipo IIb - lesão completamente plana; ᴥ» Tipo IIc - lesão levemente deprimida; ᴥ» Tipo III - lesão ulcerada; ᴥ» Tipo lla+llc - lesão levemente elevada com componente deprimido (tipo misto); ᴥ» Tipo llc + lIa - lesão levemente deprimida com elevação nas bordas ou na parte central (tipo misto). Lesões planas elevadas (IIa) e lesões sésseis (Is) podem ser confundidas. A diferenciação se faz medindo a altura da lesão com pinça de biópsia fechada, quando for maior que 2,5 mm é considerada séssil. |
Considerada quando a lesão superficialmente elevada é maior que 10 mm. São classificadas em:
ᴥ» Lesão granular homogênea (nódulos pequenos);
ᴥ» Lesão granular mista (nodulações maiores e irregulares);
ᴥ» Lesão não granular plana;
ᴥ» Lesão não granular com componente deprimido.
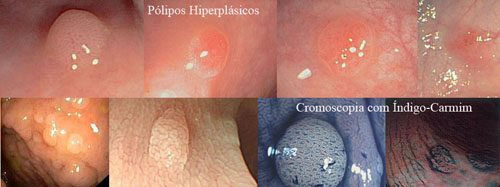
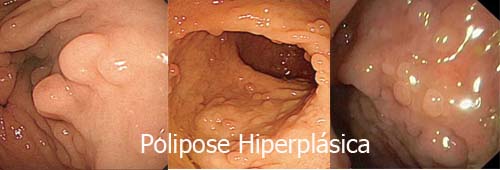
1. Hiperplásicos
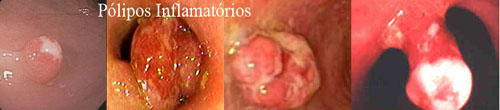
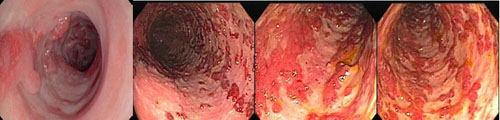
2. Inflamatórios
3. Pseudopólipos
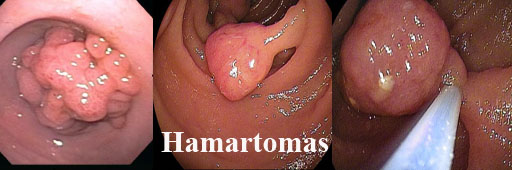
4. Hamartomas
1. Adenoma
a. Adenoma tubular.
---b. Adenoma tubulo-viloso.
---c. Adenoma viloso.
2. Serrilhado
---a. Adenoma serrilhado séssil
---b. Adenoma serrilhado séssil tradicional
---c. Misto
|
➤ Os pólipos hiperplásicos puros menores que 10 mm não têm potencial de se tornarem câncer e não necessitam de seguimento após a retirada.
➤ Os pólipos hiperplásicos maiores que 10 mm tem um comportamento semelhante aos pólipos serrilhados com risco de virar câncer e necessitam de seguimento após a retirada. |
|
Os critérios para o diagnóstico da polipose hiperplásica são:
|
|
São lesões resultantes do tecido de granulação em regeneração. Geralmente únicos e podem atingir tamanhos consideráveis e tornarem-se pediculados quando existe risco de sangramento e obstrução intestinal. Pode ter uma aparência endoscópica sinistra, irregular, avermelhado e ulcerado ou apenas regular e brancacento. Não apresentam risco de transformação maligna e não necessitam de seguimento pós-polipectomia. Quando maiores de 10 mm devem ser removidos por serem semelhantes e endoscopicamente indistinguíveis dos pólipos neoplásicos.
|
|
São ilhas de mucosa residual após cicatrização de processos inflamatórios e ulcerados no cólon e reto de longa duração. Geralmente são múltiplos de várias formas. Não apresentam risco de transformação maligna e não necessitam de ressecção e muito menos de seguimento pós-polipectomia.
|
|
São coleções de tecidos normais (compõem a lâmina própria) organizadas de modo anormal e desorganizada.
|
➀ Perda do controle de crescimento epitelial;
➁ Mitoses generalizadas em todas as camadas das criptas da mucosa;
➂ Importante alteração na renovação celular;
➃ Menor diferenciação celular;
➄ Maior produção de muco.

|
• Adenoma tubular - No mínimo 80% de sua estrutura são tubulares.
|

|
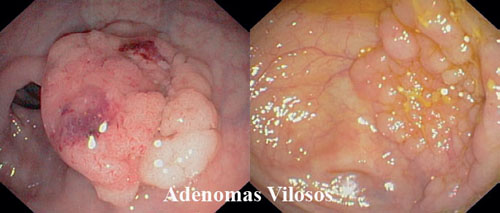
• Adenoma túbulo-viloso - Padrão misto, cada padrão representa mais de 20% de sua composição.
|
|
• Adenoma viloso - No mínimo 80% de sua estrutura são digitiformes.
|
|
Geralmente maior que 0,5 cm, plano ou séssil, localizado predominantemente à direita, podendo atingir mais de 2 cm. A lesão é difícil de detectar pela colonoscopia por geralmente ser plano ou séssil, da mesma cor da mucosa que o rodeia e o muco que o cobre apagar o seu padrão vascular. O câncer colorretal originado do adenoma serrilhado plano ou séssil apresenta histologia mucinosa, e se diferencia da Síndrome de Lynch por ocorrer em pacientes com mais de 65 anos e por ser derivado do pólipo serrilhado ao invés do adenoma.
|

|
Geralmente pediculado, exibe arquitetura complexa serrilhada, podendo ser confundido com adenoma viloso. Estes pólipos devem ser ressecados completamente sempre que possível. Apenas recentemente estudos avaliaram o risco dos pólipos serrilhados estratificando-os pelo tamanho, número, presença de displasia e morfologia. Não existem estudos de seguimento longo.
|
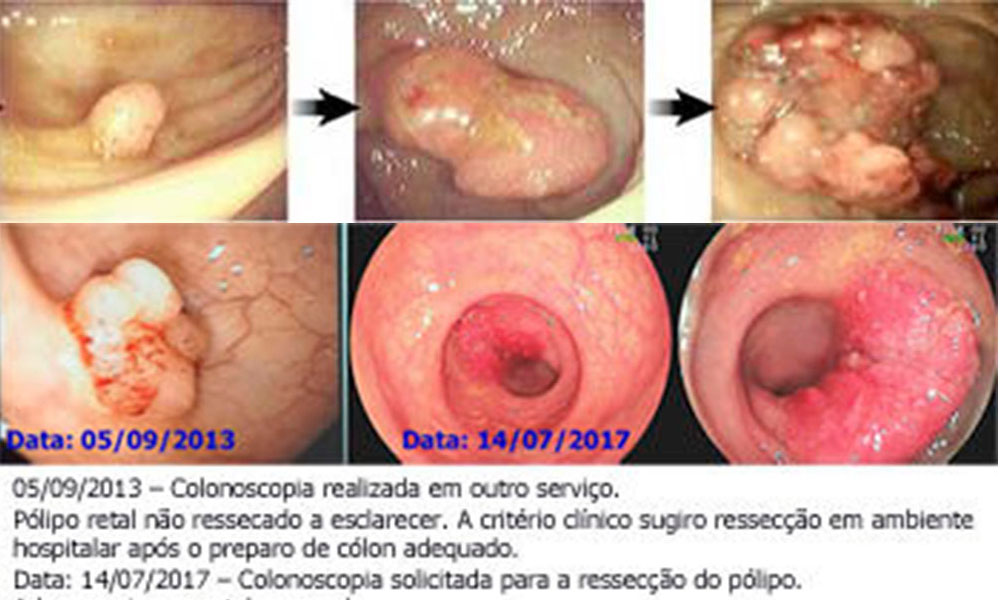
➁ O câncer colorretal surge cerca de cinco anos após o surgimento dos pólipos, isto é, em faixas etárias maiores;
➂ Cerca de um terço dos intestinos retirados com câncer colorretal incluem um ou mais pólipos associados (a frequência é seis veze s maior que a encontrada nos intestinos retirados para outras doenças;
➃ O tamanho do adenoma é o principal fator de risco para o surgimento da displasia de alto grau e o câncer.
|
➀ Os adenomas e serrilhados colorretais são encontrados em cerca de 30% das colonoscopias em pacientes acima dos 45 anos;
➁ O tamanho do adenoma é o principal fator de risco para o encontro da displasia de alto grau, que invariavelmente evolui para o câncer colorretal; ➂ Cerca de 30% dos adenomas vilosos possuem áreas de displasia de alto grau (30%), o que explica o maior risco de evoluir para o câncer colorretal; ➃ O tempo médio de transformação para o câncer é de 7 anos para os pólipos com displasia de alto grau e de 11 anos para os adenomas de baixo grau; ➄ Os estudos que analisaram a morfologia dos adenomas encontraram diferenças importantes na prevalência da displasia de alto grau e do câncer colorretal. Encontrada em 75% dos adenomas deprimidos, em comparação com 14,3% e 8,3% dos adenomas planos e polipoides, respectivamente. |

|
Responsável por 80% dos cânceres colorretais esporádicos.
O primeiro evento é a inativação do gene APC o que leva a um crescimento celular anormal e por múltiplas vias de carcinogênese, progride para carcinoma invasivo. |
❶ Recentes estudos moleculares fornecem evidências convincentes da evolução do adenoma serrilhado séssil ou plano, mais frequente no cólon direito e do adenoma serrilhado tradicional geralmente pediculado ou subpediculado, mais frequente no cólon esquerdo para o carcinoma serrilhado do cólon direito e do cólon esquerdo respectivamente. Do adenoma serrilhado misto quando existe a sobreposição de ambos e presentes tanto no colon direito quanto no cólon esquerdo;
❷ Pólipo serrilhado residual é encontrado em cerca de 5,8% dos cânceres colorretais;
❸ O risco de câncer colorretal na polipose serrilhada varia de 20% a 50%.

|
Responsável por 5% dos cânceres colorretais esporádicos. (Lancet 2000, 355(9211):1211-1214).
➊ Câncer colorretal sem componente adenomatoso, serrilhado ou “in situ”. Geralmente superficiais ou deprimidas que mesmo pequenos (≤ 10 mm) são avançados; ➋ Raros e pouco se sabe sobre as características biológicas e a história natural. Encontrado com maior frequência no oriente, especialmente no japão; ➌ Mais frequentes no cólon direito (85%), mas raramente são encontrados em pacientes pós colonoscopia (câncer de intervalo). |
ᴥ» Presença de sangue rutilante acompanhando as evacuações; ᴥ» Períodos de diarréia e/ou constipação não explicados pela dieta ou por viroses;
ᴥ» Dor em cólica na região gástrica;
ᴥ» Sensação frequente de distensão abdominal;
ᴥ» Perda de peso persistente e não explicada;
ᴥ» Astenia.
➋ Assim, os que não possuírem a mutação não mais precisarão participar do intensivo programa de rastreamento, enquanto os portadores de mutação devem seguir com o rastreamento e serem indicados para a cirurgia profilática.
➋ Dano psicológico causado pela notícia de resultado positivo no teste de DNA, podendo ser acompanhado por sentimentos de raiva, ansiedade e depressão;
➌ Discriminação genética, principalmente na área social, casamento, saúde e emprego;
➍ Confiança entre o paciente e o médico, devido ao medo da discriminação;
➎ Questões éticas, entre outras.
Aproximadamente 75 a 80% dos indivíduos com FAP têm um dos pais afetado. Portanto, sempre é apropriado avaliar seus pais com teste molecular do gene APC. Aproximadamente 20 a 25% de indivíduos com FAP têm o gene alterado como resultado de uma mutação genética nova.
O risco para os irmãos depende da condição genética dos pais. Se um dos pais é afetado, o risco é de 50%. Se nenhum dos pais de um indivíduo com FAP se encontra nos critérios clínicos para a doença, o risco de os irmãos de um indivíduo afetado terem FAP é o risco populacional para essa desordem.
Todos os filhos de um indivíduo com FAP têm uma chance de 50% de herança da mutação.
O risco para os outros membros da família depende da condição dos pais do probando. Se um dos pais encontra-se afetado, os integrantes de sua família estão sob risco.
1- Um membro seja parente em 1º grau dos outros dois.
2- Pelos menos 2 gerações sucessivas acometidas.
3- Pelo menos um dos casos de câncer colorretal diagnosticado abaixo dos 50 anos.
4- Polipose adenomatosa familiar deve ser excluída.
2- Indivíduos com 2 cânceres relacionados com HNPCC, incluindo câncer colorretal metacrônicos e sincrônicos ou cânceres extracólicos associados.
3- Indivíduos com câncer colorretal e um parente de 1º grau com câncer colorretal ou câncer extracólico relacionado com HNPCC e/ou adenoma colorretal (um dos cânceres diagnosticado abaixo dos 45 anos e os adenomas abaixo dos 40 anos).
4- Indivíduos com câncer colorretal ou endometrial diagnosticado abaixo dos 45 anos.
5- Indivíduos com câncer de cólon direito com padrão histológico indiferenciado abaixo dos 45 anos.
6- Indivíduos com câncer colorretal com células em anel de sinete diagnosticado abaixo dos 45 anos.
7- Indivíduos com adenomas diagnosticados antes dos 40 anos.
8- Cânceres extracólicos associados: endométrio, ovário, estômago, hepatobiliar, intestino delgado, células de transição da pelve renal ou ureter.
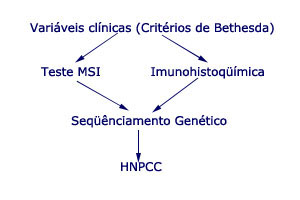
|
Embora isso pareça simples, na prática os médicos encontram enormes dificuldades para estabelecer tal diagnóstico, seja porque não dispõem de informações familiares adequadas com relação ao histórico do paciente, seja porque o sequenciamento genético não é ainda uma realidade na prática médica.
|
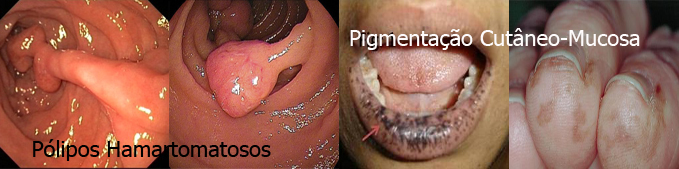
|
É uma síndrome herdada de forma autossômica dominante e se caracteriza clinicamente pela associação de pigmentação melanótica cutâneo-mucosa com pólipos hamartomatosos encontrados no trato digestivo (principalmente no intestino delgado) e, ocasionalmente, no trato urinário e respiratório. As manchas aparecem frequentemente na cavidade oral, regiões perioral e periorbitária, dedos das mãos e pés ou genitália, podendo variar em coloração.
|
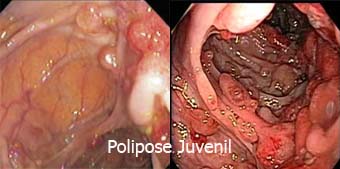
|
A Polipose Juvenil é uma síndrome autossômica dominante. É uma condição incomum e afeta de 1 em 100.000 a 1 em 160.000 pessoas e costuma se manifestar entre 4 e 14 anos de idade.
|
1- Mais de 5 pólipos juvenis no cólon ou no reto, ou;
2- Pólipos juvenis em outras áreas do trato gastrointestinal, ou;
3- Qualquer número de pólipos juvenis e uma história familiar positiva.
1- Polipose Juvenil da infância é associada a sangramento gastrointestinal de repetição, prolapso retal e intussuscepção, que ocasionam a morte precoce dos pacientes;
2- Polipose Juvenil colônica é a forma mais comum; os pólipos localizam-se no cólon e os portadores têm um prognóstico bom;
3- Polipose gastrointestinal juvenil generalizada quando a distribuição dos pólipos se assemelha à verificada na Síndrome de Peutz-Jeghers, mas a pigmentação da mucosa oral, lábios e dedos está ausente.
Os pacientes portadores da Polipose Juvenil devem ser acompanhados rigorosamente devido ao alto índice de recidiva dos pólipos.
1- Seis ou mais manchas café com leite com diâmetro superior à 5 mm em pacientes menores de 6 anos;
2- Seis ou mais manchas café com leite com diâmetro superior à 15 mm em pacientes maiores de 6 anos;
3- Sardas inguinais e/ou axilares;
4- Glioma de nervo óptico;
5- Dois ou mais neurofibromas de qualquer tipo ou um neurofibroma plexiforme;
6- Dois ou mais hamartomas de íris (nódulos de Lisch);
7- Lesões ósseas distintas como displasia do osso esfenóide e/ou afilamento da córtex de ossos longos com ou sem pseudo-artrose;
8- Parente de primeiro grau com NF1 diagnosticada de acordo com os critérios anteriormente citados.

|
É uma polipose hamartomatosa gastrointestinal generalizada associada à hiperpigmentação cutânea, perda de cabelo e atrofia ungueal. Associa-se a diarreia crônica e desnutrição com hipoproteinemia.
|
É uma doença com mau prognóstico. A maioria dos pacientes morrem por causa de um quadro de desnutrição grave, frequentemente complicado com infecções.
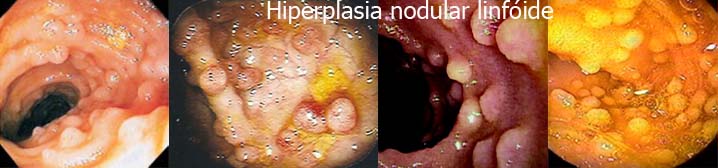
|
É uma desordem linfoproliferativa que resulta em nódulos linfóides hiperplásicos no intestino delgado, estômago e cólon, podendo estar relacionada com síndromes de imunodeficiência variadas.
|
Em pacientes com imunodeficiência associada, os nódulos estão limitados ao intestino delgado, enquanto que nos imunocompetentes, estes localizam-se também no cólon.
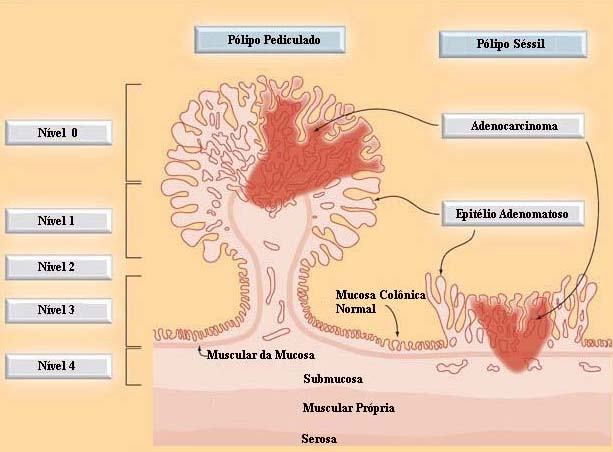
|
ᴥ» Nível 0 corresponde ao carcinoma in situ;
ᴥ» Nível 1 corresponde a invasão da muscular da mucosa limitada à cabeça do pólipo; ᴥ» Nível 2 corresponde à invasão do colo do pólipo (o colo é definido como a região do pedículo onde se dá a transição entre o componente adenomatoso e o revestimento epitelial normal); ᴥ» Nível 3 corresponde à invasão da haste ou pedículo; e ᴥ» Nível 4 corresponde à invasão da submucosa da parede intestinal na base do pólipo. As lesões sésseis com câncer invasivo restrito à submucosa (T1) e nível 4 de Haggitt são subdividido em: ᴥ» sm1 - invasão do terço mais superficial da submucosa, atingindo somente a camada muscular da mucosa; ᴥ» sm2 - invasão do terço médio; ᴥ» sm3 - invasão superficial da muscular própria. |
➤ Câncer invasivo em qualquer situação não é considerado curado e a cirurgia clássica (colectomia segmentar) está indicada na maioria porque o risco de comprometimento linfonodal chega a 9%.
➤ Carcinoma indiferenciado ou pouco diferenciado;
➤ Embolização linfática;
➤ Embolização venosa;
➤ Nível 4 de Haggitt;
➤ Nível de invasão Sm3;
➤ Margem de ressecção: menor do que 2 mm.
Em pacientes com pólipos hiperplásicos pequenos (<10 mm) confinados ao reto ou sigmóide, a colonoscopia de vigilância ou seguimento é recomendada em 5 a 10 anos, igual aos pacientes de risco médio.
O intervalo de vigilância é baseado no tamanho dos pólipos e na histologia.
2- Indivíduos com adenoma serrilhado séssil ou pediculado ≥ 10 mm, com displasia ou adenoma serrilhado tradicional são tratados como adenomas de alto risco com uma primeira colonoscopia de vigilância em três anos.
3- Outras recomendações de consenso sugeriram um acompanhamento colonoscópico mais precoce (intervalo de um a três anos) em indivíduos com dois ou mais adenomas serrilhado séssil ou pediculado ≥ 10 mm e naqueles com qualquer displasia.
1- Pelo menos cinco pólipos serrilhados proximais ao cólon sigmóide, dos quais dois ou mais são ≥10 mm.
2- Qualquer número de pólipos serrilhados proximais ao cólon sigmóide em um indivíduo que tenha um parente de primeiro grau com síndrome da polipose serrilhada.
3- Mais de 20 pólipos serrilhados de qualquer tamanho, distribuídos por todo o cólon.
Estratégias de rastreamento para pacientes com síndrome da polipose serrilhada e sua família não estão bem definidas. Pólipos ≥ 1 cm devem ser ressecados completamente. Os intervalos subsequentes da colonoscopia são baseados no número e tamanho dos pólipos, bem como no número de adenomas concomitantes, mas geralmente devem ser realizados entre um e três anos.
1- Câncer colorretal documentado ou suspeito.
2- Pólipos com displasia de alto grau ou múltiplos adenomas maiores que 6 mm.
3- Aumentos no número de pólipos em exames consecutivos.
4- Incapacidade de examinar adequadamente o cólon por causa de múltiplos pólipos diminutivos.
5- Preferência do paciente para evitar a vigilância por colonoscopia.
Pólipos adenomatosos com mais de 25% de componente viloso, displasia de alto grau ou câncer invasivo são fatores de risco para o desenvolvimento de câncer colorretal no futuro. Em alguns estudos, a vilosidade também prediz adenomas avançados no futuro.
O número de adenomas na colonoscopia e cumulativamente ao longo da vida é o fator de risco mais consistente para o câncer colorretal no futuro. Estudos sugerem que pessoas com um ou dois pólipos adenomatosos pequenos e tubulares apresentam um risco pouco aumentado de câncer colorretal no futuro. Em contraste, a presença 3 ou mais adenomas de qualquer tamanho, prediz uma taxa maior de adenomas avançados e câncer no futuro. O risco é tanto maior quanto maior o número de adenomas encontrados.
Pacientes com um ou mais adenomas ≥ 10 mm têm um risco aumentado de câncer no futuro em comparação com aqueles sem neoplasia ou com um ou dois adenomas ≤ 10 mm. O risco de neoplasia avançada aumenta com o tamanho do adenoma. Em comparação com pacientes com adenomas <5 mm, aqueles com adenomas de base de 10 a 19 mm e ≥ 20 mm têm um risco significativamente maior de câncer no futuro (8, 16 e 19 por cento, respectivamente).
➤Se um adenoma de baixo risco for detectado, a próxima colonoscopia de vigilância deve ser realizada em 3 a 5 anos.
➤Se um adenoma de alto risco for detectado, a próxima colonoscopia de vigilância deve ser realizada em 2 a 3 anos.
➤Adenoma avançado: adenoma tubular ≥ 10 mm; adenoma viloso e displasia de alto grau.
Pessoas com adenoma avançado (≥10 mm, histologia vilosa ou displasia de alto grau) ou entre 3 e 10 adenomas em sua colonoscopia devem ser submetidos a uma primeira colonoscopia de vigilância em 2 a 3 anos. Pacientes com mais de 10 adenomas devem ser avaliados para a síndrome de câncer colorretal hereditário e realizar colonoscopia de vigilância em 2 anos ou menos (veja as indicações para avaliação genética).
O momento da colonoscopia de vigilância subsequente é baseado nos achados da primeira colonoscopia de vigilância.
➧ Se não forem encontrados adenomas na primeira colonoscopia de vigilância, a próxima colonoscopia de vigilância deve ser realizada em 3 a 5 anos. Pacientes com um adenoma de alto risco em qualquer exame permanece como alto risco e devem ter intervalos de 2 a 3 anos de acompanhamento para todas as colonoscopias de vigilância subsequentes.
➧ Se um adenoma de baixo risco for detectado, a próxima colonoscopia de vigilância deve ser realizada em 3 a 5 anos.
As recomendações assumem que a colonoscopia de base foi completa e adequada e que todos os pólipos visíveis foram completamente removidos.
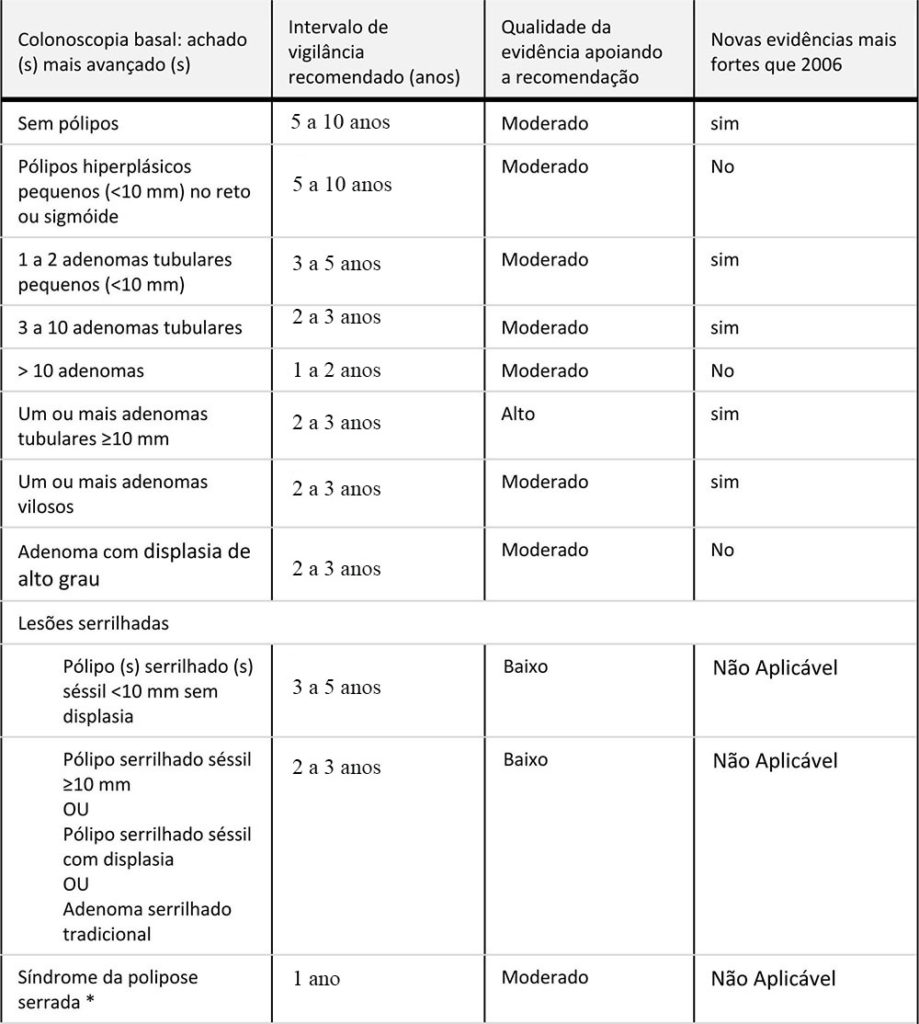
|
Recomendações para intervalos de vigilância e triagem do Câncer Colorretal em indivíduos com risco médio inicial.
|

|
Recomendações para vigilância do câncer colorretal após a primeira colonoscopia de vigilância.
|
É recomendado repetir a colonoscopia entre 6 meses a um ano. Pesquisar o provável motivo do preparo inadequado como: transgressão na dieta e/ou intolerância aos laxantes com vômitos. Caso o paciente tenha feito o preparo corretamente, a nova colonoscopia requer um preparo mais rigoroso, como o usado para o constipado grave. Veja abaixo.
1. Já fez a colonoscopia antes com preparo ruim?
2. Com que frequência vai ao banheiro evacuar? Demora mais de 4 dias?
3. Faz uso de antidepressivo?
4. Quem vai fazer a colonoscopia é diabético ou possui alguma limitação física como sequela de AVC, demência, doença de Parkinson ou uso de cadeira de rodas ou muleta/bengala?
2- Preparo para pacientes com COLOSTOMIA com o coto retal: fazer o preparo igual aos demais pacientes, mas realizar também a lavagem do coto retal com o Fleet enema ou Phosfoenema---130ml---2 unidades.
As informações contidas neste artigo são apenas para fins educacionais e não devem ser usadas para diagnóstico ou para orientar o tratamento sem o parecer de um profissional de saúde. Qualquer leitor que está preocupado com sua saúde deve entrar em contato com um médico para aconselhamento.